

Atum Crispr Grna Design Tool
CRISPR — How It Works, Top Applications and How to Use It Yourself.
![]()
CRISPR is a tool that is used to edit DNA (and, recently, RNA). I use this DNA-editing wizard on a daily basis as part of my ongoing experiments at Imperial College London; the tool has significantly expedited the pace at which I can engineer living organisms. Since late 2012, CRISPR research has exploded in a way that few scientists could have foreseen. Entire labs have arisen almost overnight that harness this powerful DNA-editing system to screen for disease and delete genes in a rapid and precise manner.
One of the be s t parts of CRISPR is its ease-of-use. A novice scientist can be up-and-running with the technology in less than one week — it is really that quick to learn. The sequences for the CRISPR-associated proteins, including Cas9 and Cpf1, can be ordered from a DNA manufacturer (IDT, GeneArt and more) or ordered as a pre-made DNA sequence (Addgene). Plasmids to express gRNAs can be designed and ordered just as easily. In rapidly growing organisms, such as E. coli, genes can be modified in under a week using the CRISPR system. This powerful DNA-editing tool is not limited to bacteria, however; CRISPR has proven effective in almost every cell line tested, from bacteria and archaea to mice and humans. Anybody, from enthusiasts to professionals, can quickly learn to use the system for a multitude of applications. In this article, I will discuss how the native CRISPR system works and how CRISPR was repurposed for utility in the laboratory. I will also provide a step-by-step guide so that you can begin using CRISPR yourself and will discuss ten modern applications of the technology. If, at the end of this article, you are still eager for more, take a look at the resources (bottom of the page).

Want to read this story later? Save it in Journal .
Am I missing important details? Is something on this page incorrect? Help me make this section better by leaving a comment!
How CRISPR Works in Nature (The Big Picture)
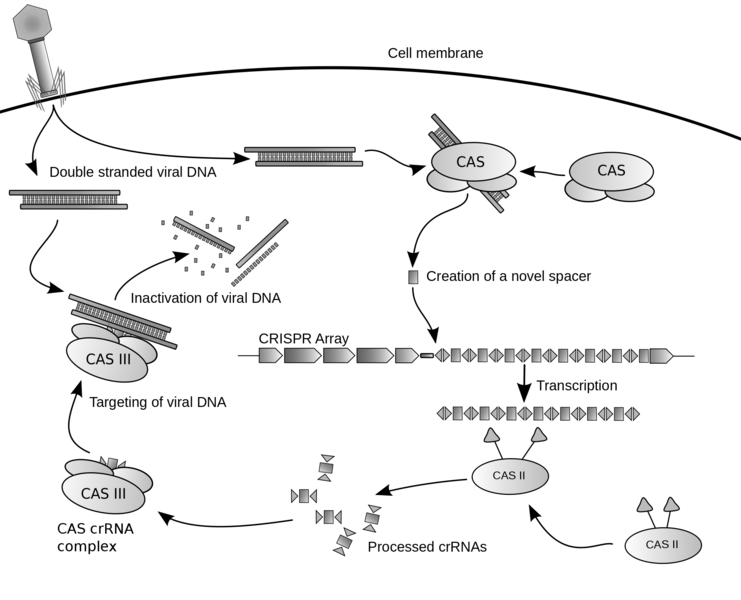
CRISPR is a modified bacterial defense system. When bacteriophages (viruses that target bacteria) attack a bacterium, they do so by injecting their DNA through the bacterial cell membrane. This bacteriophage DNA, once inside the cell, can reprogram the invaded bacterial cell and coax it into making more bacteriophages. Bacteria, being the resilient buggers that they are, have developed ingenious methods to fend off bacteriophages. One of these methods is CRISPR. Every time that a bacterial cell encounters a new bacteriophage, it cuts up a bit of the invading bacteriophage DNA and stores it in its own genome. That way, if the bacterial cell ever encounters that type of bacteriophage again, it can 'pull out' that stored piece of DNA and use it to recognize and destroy the invader. The DNA that the bacteria stores as a defense mechanism is used to fight off bacteriophages in the following way:
1. DNA from the invading bacteriophage is incorporated into a region of the bacterial genome, called the CRISPR locus.
2. During the next bacteriophage invasion, DNA (A, T, G, C) in the CRISPR locus corresponding to the bacteriophage invader is transcribed into a short piece of RNA (A, U, G, C) called a CRISPR RNA (or crRNA).
3. crRNAs bind to a large protein (called an effector complex) and guides it to the bacteriophage DNA, the sequence of which complements the crRNA sequence. The effector complex and its crRNA bind to the invading bacteriophage DNA via Watson-Crick base pairing.
Note: Some CRISPR systems require a second RNA, called a tracrRNA, which helps process the crRNA. The tracrRNA is complementary to the pre-processed crRNAs. After cleavage by RNAse III, a ribonuclease, a crRNA/tracrRNA hybrid forms and guides the effector complex to degrade the invading bacteriophage DNA.
4. The invading bacteriophage DNA is cut and can be stored in the CRISPR locus for future invasions.
Note: A common effector complex is Cas9, which stands for CRISPR associated protein 9. Cas9 binds to the crRNA and tracrRNA, which guides the Cas9 to the appropriate DNA sequence (bacteriophage DNA, in this example). Once Cas9 binds to the target DNA, it performs a double-stranded DNA cleavage.
How CRISPR Works in the Lab
The bacterial defense system known as CRISPR has been modified to expand its utility as a genome editing tool, thanks in large part to the Doudna laboratory at UC-Berkeley, the Charpentier lab at the Max Planck Institute for Infection Biology, the Zhang lab at MIT and the Church lab at Harvard, among many others. Some of their findings will be condensed here in an effort to give a general view of how CRISPR really works to edit genomes in the laboratory.
First, the cells to be engineered must produce a guideRNA, or gRNA for short. In 2012, Doudna's group published a paper in the journal Science (led by Martin Jínek) that showed that the tracrRNA and crRNA sequences from the native CRISPR system could be fused into a single RNA sequence, which they called a gRNA, that could direct Cas9 to a specific region in the genome. This turned out to be very important, since the design of gRNAs is now considered to be extremely simple and routine in the laboratory to direct Cas9 or Cpf1 to target DNA sequences.
Once a gRNA is designed and expressed in the cell of interest, an endonuclease (usually Cas9 or Cpf1) binds to it. The newly-formed Cas9 (or Cpf1) effector complex can now recognize a sequence in the genome called a protospacer-adjacent motif, or PAM.
Note: Cas9 and Cpf1 belong to a class of Cas proteins called Class 2. These are essentially large, single proteins consisting of multiple domains that are used to bind to gRNAs and then initiate double-stranded cuts in DNA. Other Cas effector complexes consist of multiple proteins.
For Cas9 used in the laboratory, the PAM sequence is typically NGG (where N represents any of the four bases). Once Cas9 recognizes and binds to the PAM sequence, it 'checks' its bound gRNA to see if there is base-pairing complementarity between the gRNA and the DNA strand. If the two are complementary to one another, a blunt, double-strand cut is initiated in the DNA sequence at a position three base pairs upstream of the 3' edge of the PAM sequence.
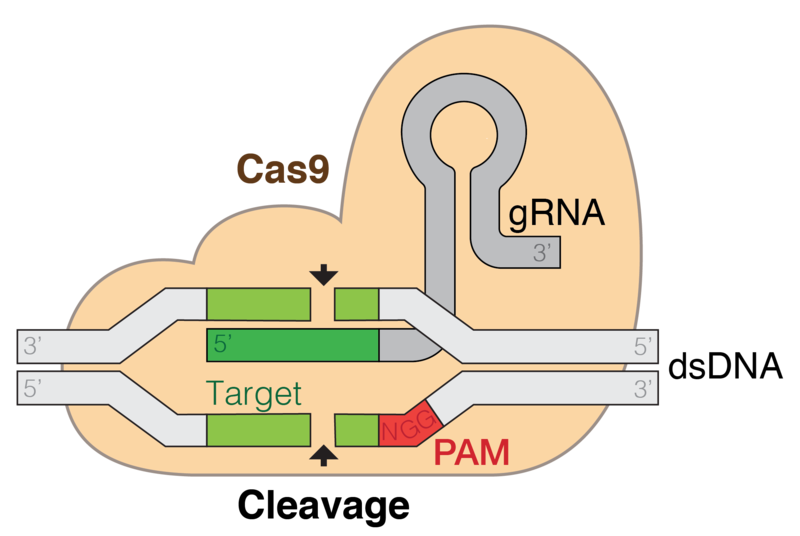
When a double-stranded DNA cut is performed, there are two ways for cells to repair the damage: non-homologous end joining (NHEJ) or homology directed repair (HDR). NHEJ is faster, but more error-prone, than HDR. When the cell uses NHEJ to repair the damage, it often results in nucleotide insertions at the DNA strand break — this means that some cells have permanently damaged DNA that expresses a mutant, non-functional gene. This means that, when CRISPR-Cas9 is used to generate double-strand breaks in DNA in a large population of cells, some of the cells will misrepair the damage and have permanent loss-of-function mutations in the targeted gene. This is a researcher's dream to quickly and efficiently delete genes in organisms.
Long story short: 1) Decide which gene you want to cut. 2) Design a gRNA to target a specific PAM sequence near that region. 3) Express that gRNA in the cell of interest in addition to an endonuclease protein such as Cas9 or Cpf1. 4) Voila! The DNA will be cut at that position and some of the resulting cells will have loss-of-function mutations in the target gene.
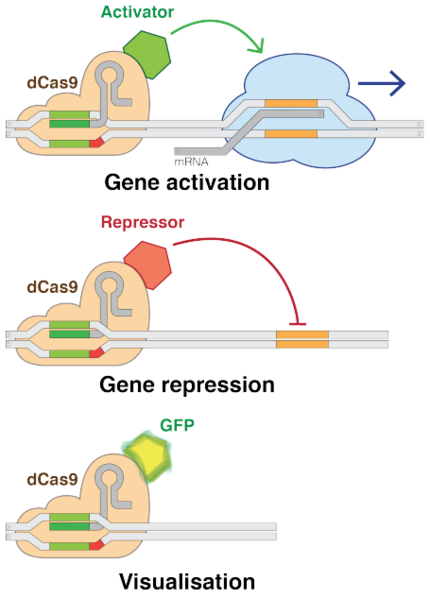
Note: CRISPR can be used for more than just DNA editing. By modifying just two amino acids in the Cas9 and/or Cpf1 proteins, these enzymes become mutants that lack the ability to cut DNA. Instead, they simply bind to the targeted DNA sequence and can be programmed to either activate or repress gene expression at that position in the genome. These proteins are called nuclease-deficient endonucleases.
Am I missing important details? Is something on this page incorrect? Help me make this section better by leaving a comment!
Step-by-Step Guide on Using CRISPR:
It is incredibly simple to use CRISPR in the laboratory. Though there are many methods to do so, the guide below will demonstrate how to select a gene to target, design a gRNA to delete that gene, and then actually delete it in living cells.
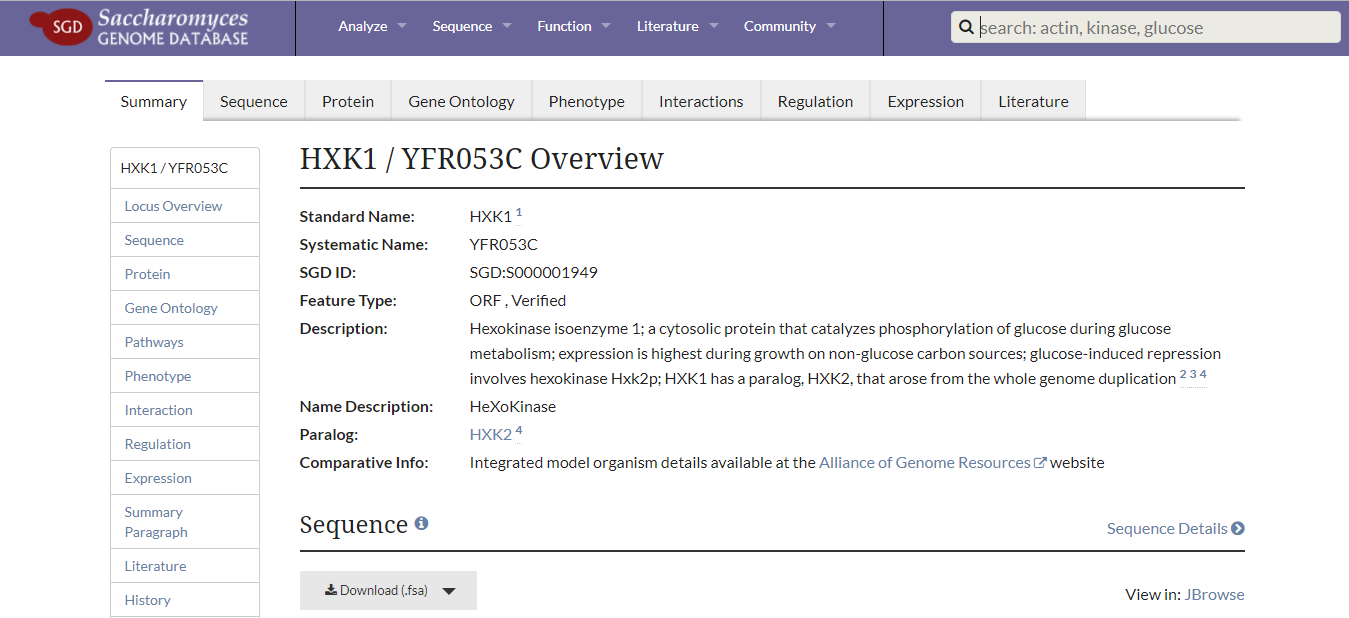
1. Decide which gene to modify (cut, activate or inhibit). For this example, we will knockout the hexokinase gene in Saccharomyces cerevisiae. This is a commonly-utilized species of yeast — the exact same strain that is used to brew beer. Hexokinase is an enzyme that phosphorylates glucose as part of the cellular process called glycolysis. By removing hexokinase, yeast cells will have impaired glycolytic flux.
First, retrieve the DNA sequence of hexokinase using the Saccharomyces Genome Database. Simply search for any gene of interest and select it from the drop-down — HXK1 is the gene acronym for isoenzyme 1 of hexokinase.
From this page, download the .fsa file containing the sequence of interest. It may be useful to download the Genomic DNA +/- 1kb because this will provide the actual coding sequence of the gene of interest in addition to upstream and downstream DNA regions. Save the file to your computer.
2. Decide which endonuclease protein to use. There are more enzymes than just Cas9 which can be used for genome engineering. Cpf1 is another Class II endoribonuclease that has been repurposed for genome engineering. For this example, Cas9 will be used because it is easy to work with and possesses all of the functionalities necessary to create a DNA break in a single protein.
Note: Nuclease-deficient Cas9 or Cpf1 can function as a gene activator or gene repressor by fusing it to an Activation Domain or Repression Domain. Common activation domains include B42, VP16 and VP64, while common repression domains include KRAB and Mxi1. For the purposes of this example, the activation and repression domains will not be necessary, as the goal is simply to create a double-stranded DNA break in the hexokinase gene in Saccharomyces cerevisiae cells. The following steps are all the same, however, regardless of whether you would like to activate gene expression or edit a target gene — the only difference is the endonuclease used.
3. Design the gRNA to target the gene of interest. Benchling is one of many useful websites that offers a free, built-in gRNA design tool. Other options include: GenScript, E-CRISP, ATUM and MIT's CRISPR Design. To design the gRNA, first upload the target gene of interest (hexokinase, in this case) into Benchling. To do this, select Inventory and then click on the + icon in the upper right-hand corner of the left sidebar. Click Import DNA sequences and then drag-and-drop the .fsa file to the pop-up screen. After a green bar appears that reads Upload done, close the window. The gene sequence will now automatically open onto the main center column in the browser.
In the middle column of your screen, the DNA sequence of the imported .fsa file (A, T, G and C) will appear, along with a series of numbers, in increments of ten, beneath them. Since the Genomic DNA +/- 1kb was used in this example, this sequence includes one-thousand base pairs of DNA before the start of the hexokinase gene in addition to one-thousand base pairs of DNA at the end of the gene sequence.
To design a gRNA to cut this gene, click the icon on the right-hand side of the screen. This is the CRISPR menu. Select Design and Analyze Guides. This will pull up a new screen entitled Design CRISPR Guides: Guide Paramaters. In this menu, ensure that Single guide is selected. Guide length for Cas9 can remain at 20. Select the genome of the organism that you are using; in this case, the R64–1–1(SACCER3, SACCHAROMYCES CEREVISIAE) genome, with a PAM sequence of NGG. Advanced settings can be left unchanged. If desired, the Guide Composition Settings under Advanced Settings can be modified. One common change is to increase the desired G+C% of the designed gRNA, as guanine and cytosine bases form more stable bonds with the target DNA strand, but this is not necessary for this example. Click Finish.
Now a new column will appear on the right-hand side of the screen. In the Target Region windows, enter the beginning of the region of DNA which you would like to target for Start and the end of the region of DNA to target for End. For this example, the values of 1100 to 1600 were chosen because they fall within the hexokinase coding sequence, but are otherwise arbitrary. Once these numbers are entered, click the green Create button.
The screen will now populate with a multitude of double-wide boxes with numbers inside of them. These are indicating the On-Target and Off-Target scores of the gRNAs. These scores range from 0–100, with 100 being the 'best' in both cases. The On-Target score is generated based on experiments published by John G. Doench and Nicolo Fusi in 2016, in which they tested the efficacy of thousands of gRNAs to determine rules for predicting the best gRNA designs. A high On-Target score indicates that the gRNA will bind well near a particular PAM sequence. A low Off-Target score suggests that there are DNA sequences similar to your target gene elsewhere in the genome and, consequently, the gRNA could bind to multiple DNA sequences, resulting in decreased efficiency.
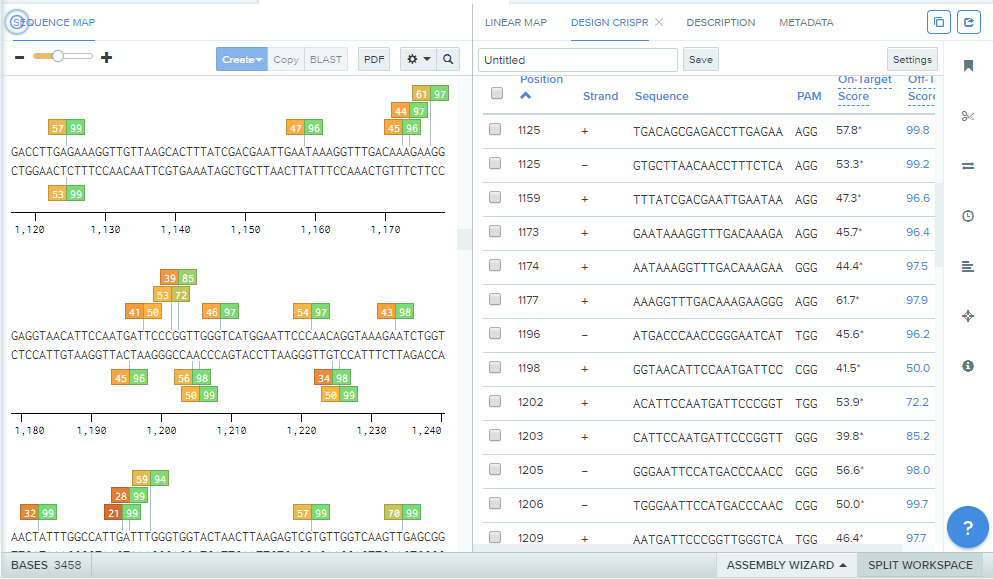
To get accurate Off-Target scores in Benchling, it is necessary to select the Genome Region of the gene that is being targeted (hexokinase, in this example). To do this, click on No Region Set underneath Genome Region. A dialogue screen will appear. Click on the Find Genome Matches button and the nifty Benchling tool will automatically predict where the gene entered (hexokinase) is from in the yeast genome.
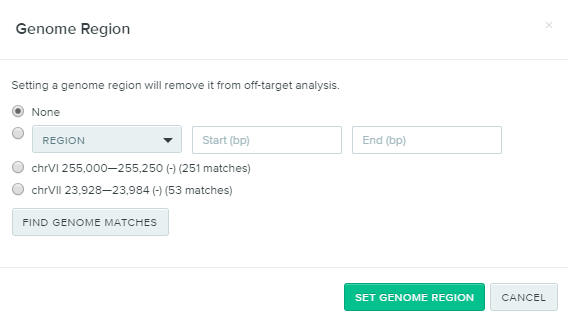
In this case, two genomic regions appear; one on chromosome 6 and another on chromosome 7. The hexokinase isoenzyme 1 that was selected for this example is on chromosome 6 at positions 255,049–253592, so the first option is the one that should be selected. The second option, chromosome 7, is another hexokinase, called hexokinase isoenzyme 2.
Click on the appropriate option for the gene of interest and hit Set Genome Region. This will update the off-target scores for the gRNAs. Now just select a gRNA in the desired region with a good On- and Off-Target score. For this example, a gRNA at position 1297 in the entered hexokinase gene sequence, with the sequence GTCGTGTTGGTCAAGTTGAG was selected due to its high On- and Off-Target scores (70.2 and 99.8, respectively). Once the desired gRNA (or gRNAs) is selected, click Assemble.
4. Assemble the gRNA Expression Vector in your browser. After selecting the desired gRNA and clicking on Assemble, a new page will appear on the right-hand side of the browser. This is where things can get a bit more complicated because different expression vectors (or plasmids, the bits of DNA that actually produce the Cas9) differ based on the organism that is being engineered. Since S. cerevisiae is being genetically modified for this example, a specific expression vector that has been optimized for yeast should be selected. By clicking on Choose Vector, a few different plasmid options will appear. The vectors on this list were provided by Feng Zhang's laboratory at MIT. All of the plasmids in the list are different — some express nuclease-deficient Cas9 (for gene activation or repression, as discussed previously) while others are optimized for CRISPR editing in mammalian cells. For the purposes of this demonstration, a new plasmid will be uploaded which has been optimized for yeast.
First, click on Upload a new plasmid and then drag-and-drop a DNA sequence file. Addgene is a non-profit plasmid repository that contains sequences for several thousands of plasmids that express Cas9 or other endonucleases. In this example, the plasmid pML107 from John Wyrick's laboratory will be used. This plasmid expresses Cas9 and contains a 'slot' to express the gRNA that was selected in Step 3. Under Sequence Information, click on Depositor Sequences: Full. A new page will appear with the full DNA sequence of the plasmid. Click on GenBank, which will download a .gbk file type that is compatible with Benchling. Drag and drop this plasmid file to Benchling.
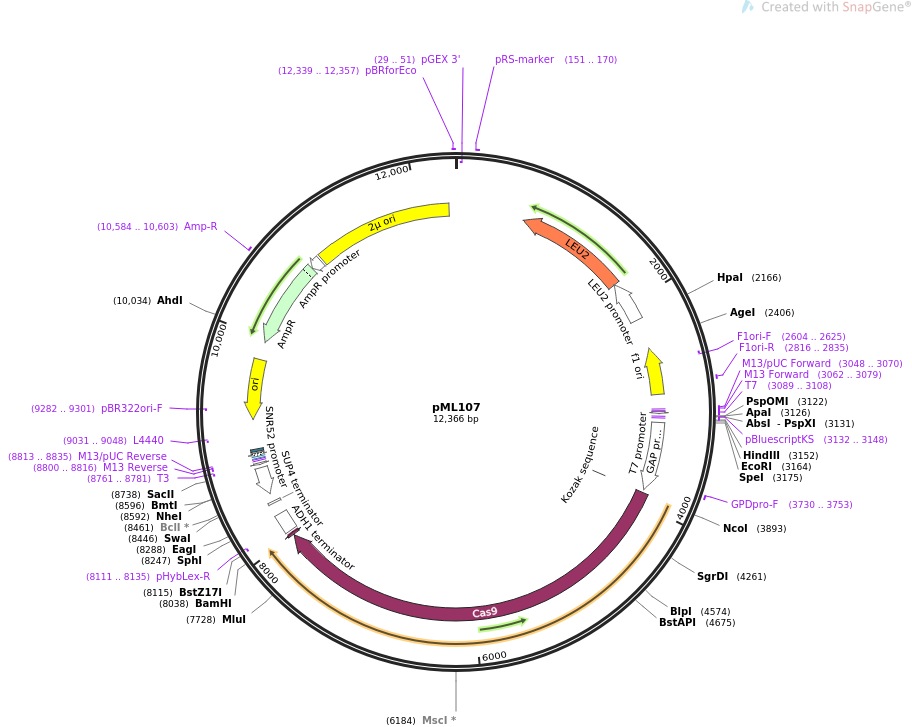
In Benchling, feel free to provide a Sequence Name for the plasmid that was uploaded. Next, select a Folder within Benchling to save the Assembled gRNA expression vector. First, however, it is necessary to instruct Benchling as to where in the uploaded plasmid sequence the gRNA should be inserted. This process relies on restriction enzymes, which you can read more about here. By uploading the plasmid sequence into Benchling (in a separate window), it is simple to analyse the DNA sequence and identify restriction sites that could be used to insert the gRNA. Oftentimes, the plasmid page on Addgene will also specify the location within the plasmid to insert the gRNA. Once this information is entered into the Benchling window, click Assemble. The constructed gRNA and Cas9 expression plasmid will now be saved to the specified folder and will also appear in the center column as a DNA sequence.
5. Assemble the plasmid at the bench! To do this, the following materials are required:
a. Expression vector — a plasmid that produces Cas9 and the designed gRNA. Addgene lists prices on the webpage for each plasmid; pML107 costs $65. The plasmid is sent via a tube that contains living bacteria. The bacteria must be destroyed and the plasmid isolated via a miniprep or chloroform:phenol extraction. Instructions on performing each of these experiments can be found online.
b. The gRNA designed by Benchling or another web-based tool. DNA is incredibly cheap to order. There are many DNA manufacturers, including IDT and GeneArt. IDT even has direct ways to order gRNAs for CRISPR editing on their webpage. Ordering a gRNA (DNA sequence) costs less than $10. Ensure that, when ordering the gRNA, you include restriction sites for its insertion into the expression vector.
c. Restriction enzymes to cut the expression vector and the gRNA and generate overlapping DNA sequences for their assembly. New England Biolabs is the world leader in restriction enzymes, offering hundreds of different options along with optimized buffers. Most restriction enzymes cost under $70 and buffers are even cheaper.
d. Miscellaneous reagents. Other enzymes, from DNA ligase (to repair the expression vector and gRNA after they are joined) and buffers (to maintain stable environments for the DNA to be manipulated) to plastic tubes and pipettes, are also required.
e. Cells to engineer. For this example, S. cerevisiae must be acquired. Fortunately, cells are absurdly inexpensive or, usually, completely free.
Once all of the necessary reagents are in place, use the restriction enzymes to cut the gRNA and the expression vector and then join the two pieces of DNA with ligase enzyme. To see detailed instructions to perform cloning in molecular biology, Khan Academy is a great resource.
6. Engineer the Cells! After creating the expression vector (which now expresses both Cas9 and the designed gRNA), it is time to get the expression vector inside living cells. This process is simpler than one might imagine and is a process called transformation. Bacteria are easier and faster to transform than eukaryotic cells.
To transform S. cerevisiae, follow this quick, intuitive protocol online. To achieve higher efficiency of transformation, a more in-depth transformation protocol is likely required. These can also be found online.
After conducting the protocol and incubating the transformed yeast at 30⁰C, it takes about two days for yeast colonies to appear. These colonies can be screened and tested using a variety of methods — a few of the colonies will be knockouts of hexokinase (or the gene that was selected), though experiments should be performed to validate that this is the case.
Top 10 CRISPR applications:
1. Pathogen diagnostics.
A team of scientists at MIT are utilizing CRISPR to detect pathogens quickly and cheaply. Led by Professors James Collins and Feng Zhang, a new CRISPR system that targets RNA, rather than DNA, called SHERLOCK (Specific High-sensitivity Enzymatic Reporter unlocking) is being used to detect viral and bacterial sequences. This technology has been used to differentiate between African and American strains of the Zika virus and can even detect genes involved in antibiotic resistance from a multitude of bacterial strains.
2. Cancer screening.
Scientists at the Dana-Farber and Boston Children's Cancer and Blood Disorders Center in Boston are using CRISPR-Cas9 to screen thousands of genes implicated in tumor-formation in mice. By removing fibroblasts from mice and deleting every gene in the genome one-by-one in these cells (by using gRNAs designed to target each gene in parallel), scientists can gather astounding amounts of data that paint a picture of which genes are involved in cancer and how they can be targeted by pharmaceuticals.
3. Metabolic engineering and manufacturing of high-value compounds.
Jay Keasling's laboratory at UC-Berkeley is internationally-renowned in the field of metabolic engineering, a discipline that rewires the metabolism of organisms to produce high-value chemicals and biofuels from cheap starting materials, such as sugars. Jay Keasling's group has recently begun using CRISPR with nuclease-deficient endonucleases (Cas9 and Cpf1 that have lost the ability to cut DNA, and instead activate or repress gene expression) to rewire organism metabolism in a manner that is significantly faster than methods used previously.
4. Mouse models/gene deletions.
CRISPR has made gene knockouts easier to make than ever before, and the technology is already being implemented to generate designer mice that lack specific genes. These animals are useful models to study disease progression for complex human illnesses, including Alzheimer's, diabetes and cancers. Jackson Laboratories, one of the world leaders in mouse genome editing, describes three easy methods for creating mice with gene deletions on their website.
5. Correction of deleterious mutations in embryos.
In 2017, it was officially reported that CRISPR had been used to edit a human genome in the United States to correct Hunter syndrome in a 44-year-old patient, though China has been using the technology to edit human embryos since much earlier (first reported April 2015). This year, CRISPR editing human patient trials will officially begin in Europe, spearheaded by CRISPR Therapeutics, a company based in Cambridge, Massachusetts.
6. CRISPR genome editing for improved animal breeding.
CRISPR-Cas9 can be used to modify DNA with unprecedented specificity and precision, allowing farmers to breed animals that possess desirable traits significantly faster than conventional breeding programs. There are numerous ethical concerns related to the genetic modification of livestock for consumption and the patent applications surrounding the use of CRISPR for livestock engineering are still actively debated.
7. Gene drives to eradicate malaria.
The Bill & Melinda Gates Foundation has, for many years, led an international effort to develop gene drives that can eradicate parasite-carrying mosquitoes. Only female mosquitoes of certain species carry the malaria-causing plasmodium. First, female mosquitoes are engineered to carry the gene drive on one of its chromosomes'. When this female is released into the population and breeds with a male mosquito, the gene drive is delivered to the offspring (only 50% of offspring will inherit the gene drive). After several generations, the gene drive can spread quickly throughout a population. To learn more about how CRISPR-Cas9 gene drives work, check out this animation from the Wyss Institute at Harvard University.
8. Modification of traits in living organisms (including butterfly wings).
Since CRISPR can be used to activate or repress gene expression, it can be used to flip gene switches 'on' and 'off' at selected times during development. Researchers have exploited this capability to control the development of wing patterns in butterflies. The study, published in the Proceedings of the National Academy of Sciences, revealed a gene master switch that controls pattern formation in butterflies and which may also control developmental processes in other organisms.
9. Cell therapy; most notably CAR T-cell therapies.
CRISPR is being used to enhance cancer immunotherapy. Several laboratories have used the technology to engineer T-cells extracted from patients with cancer. By placing specific genes into the T-cells, they can be 'programmed' to hunt down and attack tumors within the patient. A handful of companies have emerged in this space; most recently, a company called Cell Design Labs (founded by Wendell Lim, Professor of Cellular and Molecular Pharmacology at the University of California, San Francisco) was acquired by Gilead Sciences and Kite for up to $567 million.
10. Bringing Back the Woolly Mammoth!
George Church and his laboratory at Harvard are internationally-renowned; Church has played a hand in shaping almost everything related to modern DNA sequencing and synthesis technologies. He is perhaps most famous (at least in the media), however, for his aims to bring back the woolly mammoth. DNA editing technologies have already been used to bring back extinct species, such as the Pyrenean ibex, but the woolly mammoth is a much tougher problem partially because, over thousands of years, DNA degrades. Consequently, the extracted DNA from woolly mammoth hair, skin and bones needs to be repaired before it can be inserted into a viable embryo. CRISPR will simplify and streamline this process.

Am I missing important details? Is something on this page incorrect? Help me make this section better by leaving a comment!
How CRISPR Has Improved in the Last Two Years:
Here are the top five updates to the CRISPR system, each of which has mitigated essential weaknesses present in the original CRISPR gene editing system.
1. Broader PAM specificity. A major limitation of CRISPR is its reliance on PAM sequences — an endonuclease:gRNA complex can only bind near a PAM sequence (for Cas9, this is NGG, where N represents any of the four DNA bases). A recent paper in the journal Nature by the laboratory of David Liu mitigates this limitation. They evolved a new form of the Cas9 protein which has broader PAM specificity, thus enabling researchers to target gRNAs to an ever-expanding number of sites within the genome.
2. Thermostable Cas9. Though not essential for the functioning of CRISPR, it would be useful to have endoribonucleases, such as Cas9 and Cpf1, that are thermostable — that is, they remain active even at high temperatures. This is precisely what Jennifer Doudna's laboratory at UC-Berkeley accomplished in 2017. They even showed that this new form of Cas9 was more stable in human blood plasma, thus increasing its efficacy as a potential gene editing tool in humans.
3. Multiplexing gRNAs. One major weakness of CRISPR systems currently is that gRNAs are typically expressed one at a time, limiting the system to a single modification per cellular generation. To make hundreds, or even thousands, of genetic modifications to a cell, modern CRISPR systems would still take quite a long time (though not nearly as long as historical molecular biology methods). One way to address this deficiency is to multiplex gRNAs — that is, find ways to express many, many gRNAs that target multiple regions of the genome in a single cell simultaneously. There are a few ways to multiplex gRNAs; a common method today is intron splicing, wherein gRNAs are expressed from a single promoter and each gRNA is flanked by intron sequences. Most eukaryotic cells will then cleave out these intron sequences, releasing the gRNAs. Researchers have not managed to express more than a few gRNAs from a single locus, but developments in this research area are leading to rapid improvements for the CRISPR system.
4. CRISPR-Editing of RNA. CRISPR systems target DNA. But what if we wanted to make edits to RNA? This is exactly what laboratories at MIT have accomplished with the new SHERLOCK system. The laboratories of James Collins and Feng Zhang have patented the technology, which uses a new endonuclease, called Cas13, that can be used to detect both DNA and RNA. The system was published in February of 2018 by lead authors Jonathan Gootenberg and Omar Abudayyeh in the journal Science. Use of CRISPR systems on RNA holds as-yet untapped potential in pathogen diagnostics, circuit design and implementation in living cells, and much more.
5. Tri-functional CRISPR systems. Multiple CRISPR systems can be employed simultaneously within the same cell — by expressing numerous endonucleases and gRNAs at once, many different 'functions' can be carried out in the cell rapidly and specifically. By expressing Cas9, nuclease-deficient Cas9 and nuclease-deficient Cpf1 in yeast (along with three gRNAs, one for each of the endonucleases), Huimin Zhao's laboratory at the University of Illinois (Urbana-Champaign) managed to activate, inhibit and delete three unique genes at once inside of cells. There are, as yet, no limitations as to the number of endonucleases and gRNAs that can be expressed, but scientists are rapidly exploring the theoretical and experimental limits!
Am I missing important reading materials? Are there any sources that you recommend? Help me make this section better by leaving a comment!
Where to Learn More About CRISPR:
If you did not understand everything in this article, do not fret. Any gaps that may exist in your knowledge can be blamed on my explanations and not your aptitude. Fortunately, there are dozens of great resources to learn more about CRISPR, its applications, history or future. I have included a few quality resources here to aid you on your gene-editing journey!
1. A Crack in Creation: The New Power to Control Evolution by Jennifer A. Doudna and Samuel Sternberg. This recent book (published just last year) is probably the best place for an enthusiast to learn more about CRISPR. The book is written towards a general audience and explains its history, development and applications.
2. The new frontier of genome engineering with CRISPR-Cas9 by Jennifer A. Doudna and Emmanuelle Charpentier. This is an article in Science magazine that does an excellent job of explaining CRISPR, comparing it to tools that had been used previously to edit DNA. The article is tailored to scientists or CRISPR enthusiasts. It doesn't get better than hearing about CRISPR from two of the founders!
3. The Addgene CRISPR Guide. This is probably the definitive CRISPR guide — it covers pretty much everything related to CRISPR, including how to use it, designing guides, choosing an endonuclease, and everything else. Addgene, the non-profit plasmid repository that I wrote about earlier in this article, offers unparalleled resources for CRISPR systems. The articles are written for scientists but, with enough background, you will have no trouble understanding these Addgene resources.
4. CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular Biology by New England Biolabs. A detailed, but brief, guide on CRISPR, with a particular emphasis on the molecular mechanisms behind CRISPR-Cas9 (for example, how the DNA double-stranded cut is made and repaired by the cell). The article also includes some experiments that researchers can use to test their CRISPR systems.
5. CRISPR-Cas: A Laboratory Manual by Jennifer A. Doudna and Prashant Mali. This textbook covers nearly every use of CRISPR with detailed, step-by-step instructions on using the technology to edit DNA, activate or repress genes, and many other applications. This is a definitive experimental guide to understand and apply CRISPR.
Atum Crispr Grna Design Tool
Source: https://nikomccarty.medium.com/almost-everything-you-should-know-about-crispr-how-it-works-top-applications-and-how-to-use-it-61e27b04bea6
Posted by: franklinsart1949.blogspot.com
0 Response to "Atum Crispr Grna Design Tool"
Post a Comment